
An der Friedrich-Alexander-Universität Erlangen-Nürnberg gibt es die Möglichkeit, das Wahlpflichtpraktikum in der Computerchemie zu absolvieren. Studierende lernen, ihre eigene wissenschaftliche Arbeit anzufertigen und wie biologische Systeme mittels Simulationen auf atomarer Ebene untersucht werden können. Wie läuft das Praktikum ab?
Das Wahlpflichtpraktikum bietet den Pharmaziestudierenden die erste (und oft einzige) Möglichkeit, im Rahmen des Studiums einen Einblick in den wissenschaftlichen Betrieb zu erlangen. Die Inhalte des Pharmaziestudiums sind meist klar vorgeschrieben und lassen wenig Spielraum, den eigenen Interessen zu folgen. Im Wahlpflichtpraktikum hingegen hat man die Chance, sich mit einer Fachrichtung intensiv zu befassen und herauszufinden, ob Wissenschaft etwas für einen ist oder nicht.
Häufig wird das dreiwöchige Wahlpflichtpraktikum dabei in einer der Arbeitsgruppen der eigenen Universität während der Semesterferien abgeleistet, allerdings kann es auch bei Arbeitsgruppen an anderen Universitäten oder im Ausland durchgeführt werden. Durch das Praktikum bekommen Pharmaziestudierende einen aussagekräftigen Eindruck davon, wie tagtäglich geforscht wird und dass hinter der mythischen Kulisse des Labors auch nur mit Wasser und Ethylacetat gekocht wird. Zudem lernt man in den Kaffeepausen die ehemaligen oder künftigen Assistenten als Kollegen kennen und stellt fest, dass diese auch nur gewöhnliche Menschen sind (und keine fiesen Monster).
Wahlpflichtpraktikum in der Pharmazeutischen Chemie ohne organische Synthese
Besonders die Pharmazeutische Chemie bietet für gewöhnlich viele Plätze an und lockt mit spannenden
Projekten. Bei Pharmazeutischer Chemie denken viele in erster Linie an die klassische organische Synthese. Aber auch die computergestützte Wirkstoffentwicklung (CADD) mit Methoden wie Docking und molekulardynamischen (MD) Simulationen ist fester Bestandteil dieser Disziplin [1]. Trotz großer Erfolge [2, 3] hat die computergestützte Wirkstoffentwicklung aber noch nicht wirklich Einzug ins Curriculum des Pharmaziestudiums gefunden – so haben wohl die meisten im Laufe des Pharmaziestudiums Paracetamol oder ähnliche Verbindungen synthetisiert, aber die wenigsten ein Docking-Experiment durchgeführt. An der Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) versucht man, diese Lücke im Curriculum durch ein Wahlpflichtpraktikum in der Computerchemie zu schließen. Dafür werden explizit Ressourcen durch das Zentrum für Nationales Hochleistungsrechnen Erlangen (NHR@FAU) zur Verfügung gestellt, die es den Studierenden ermöglichen, eigenständig Kalkulationen und Rechnungen auf einem High-Performance-Computing-Cluster durchzuführen.
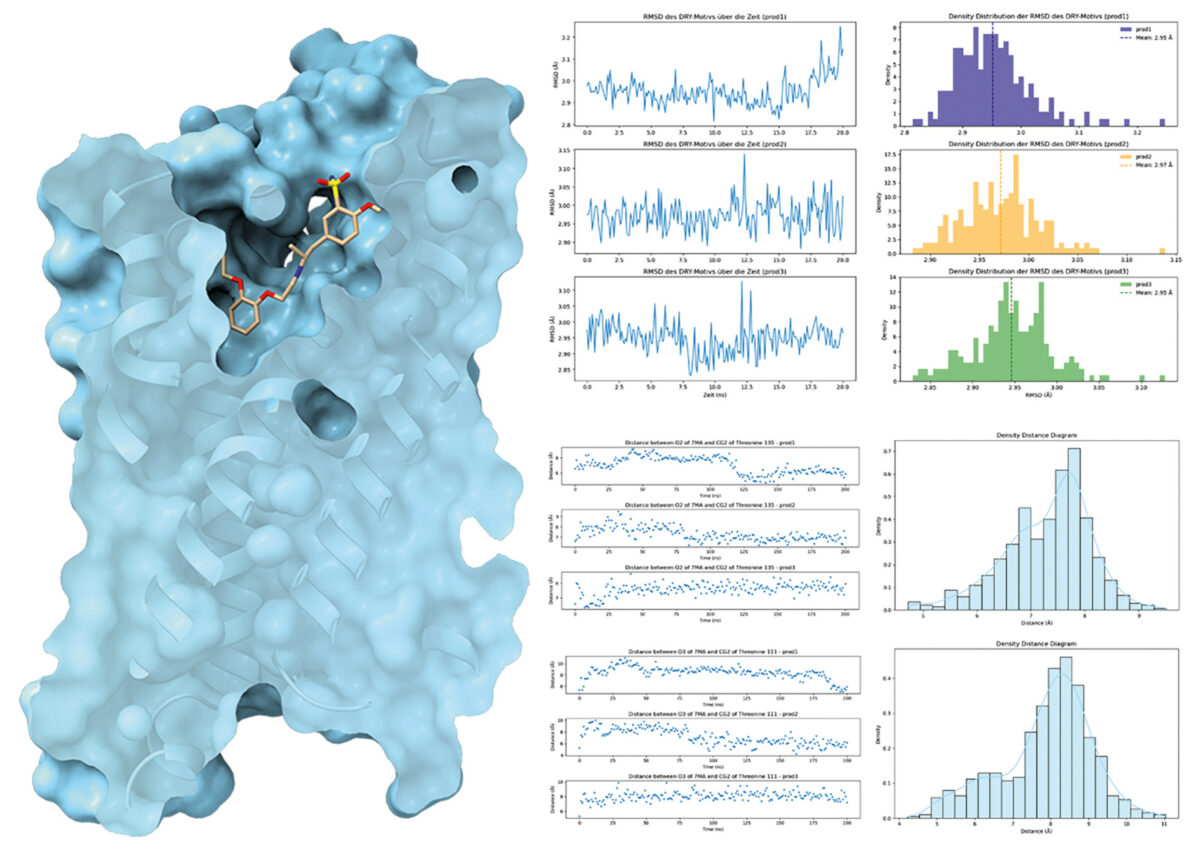
Rechts – Linien-, Streuungs-, Histogramm- und KDE-Diagramme. Diese Abbildungen wurden von Studierenden während des Wahlpflichtpraktikums erstellt.
Erforschung von GPCRs …
Das Wahlpflichtpraktikum der Computerchemie wird von der Arbeitsgruppe um Prof. Dr. Peter Gmeiner veranstaltet. Das Gmeiner-Labor forscht an G-Protein-gekoppelten Rezeptoren (GPCRs) und war an der Strukturbestimmung des β2-Adrenozeptors beteiligt [4]. Für diese und weitere Leistungen wurde Brian K. Kobilka, ein langjähriger Kollaborationspartner des Gmeiner-Labors, 2012 mit dem Nobelpreis für Chemie ausgezeichnet. Schätzungsweise wirken 30 bis 40% aller zugelassenen Medikamente über GPCRs.
… in drei Wochen
Konkret sollen die Studierenden während des Wahlpflichtpraktikums eine eigene wissenschaftliche Arbeit anfertigen. Zu Beginn des Praktikums wird jedem ein pharmakologisch relevanter GPCR zugeteilt.
In der ersten Woche des Praktikums findet ein Crashkurs zum wissenschaftlichen Arbeiten statt. Dabei lernen die Studierenden, eine Literaturrecherche durchzuführen und mit Programmen zur Literaturverwaltung (EndNote und Mendeley) umzugehen. Zudem lernen sie die Programme PyMOL und Chimera kennen, mit denen die dreidimensionalen Strukturen von GPCRs visualisiert werden können. Diese Strukturen werden aufwendig von Wissenschaftlern auf der ganzen Welt mittels Kryoelektronenmikroskopie oder Röntgenkristallografie bestimmt. Zusätzlich finden Seminare statt, in welchen die Funktionsweise von GPCRs und deren nachgeschalteten Signalkaskaden weiter thematisiert wird. Die Studierenden sollen sich in dieser Zeit damit vertraut machen, wissenschaftliche Veröffentlichungen zu finden und diese effektiv zu lesen. Alle Unterrichtseinheiten werden aufgezeichnet und anschließend auf das interne Videoportal der FAU (fau.tv) hochgeladen, damit die Inhalte in Ruhe nachgearbeitet werden können.
In der zweiten Woche loggen sich die Studierenden auf dem High-Performance-Computing-Cluster ein und bekommen die Grundlagen im Umgang mit dem Betriebssystem Linux beigebracht. Im Gegensatz zum Windows-Betriebssystem, das, wie der Name sagt, über Fenster bedient wird, wird das Linux-Betriebssystem größtenteils über die Kommandozeile mit Befehlen und Skripten bedient. Das mag zu Beginn für einige Studierende etwas befremdlich wirken und den Eindruck erwecken, man habe sich in eine Informatikvorlesung verirrt. Doch diese anfängliche Berührungsangst weicht schnell und spätestens, nachdem man feststellt, dass 100 Dateien mit einem einzigen Befehl umbenannt werden können statt mit zig Mausklicks. Anschließend modellieren die Studierenden ihr System mit dem Webserver CHARMM-GUI. Dafür wird der Rezeptor-Ligand-Komplex in eine Zellmembran modelliert, in Wasser gelöst, und eine physiologische Ionenkonzentration geschaffen. Die so generierten Dateien dienen als Startpunkt für die anschließende molekulardynamische Simulation, welche mit dem Programm GROMACS durchgeführt wird. Diese Simulationen ermöglichen es, biologische Systeme auf atomarer Ebene zu untersuchen, mit einer räumlichen und zeitlichen Auflösung, welche bis jetzt jede verfügbare experimentelle Methode übertrifft. Dafür werden die Koordinaten und Geschwindigkeiten der Atome im System alle zwei Femtosekunden berechnet, basierend auf den inter- und intramolekular wirkenden Kräften. Dadurch entsteht eine Trajektorie – vergleichbar mit einem kurzen Film, die den „Tanz der Atome“ abbildet und verdeutlicht, dass die Interaktion von einem Medikament mit einem Rezeptor dynamisch ist. Außerdem wird dabei ersichtlich, dass GPCRs eine Vielzahl von unterschiedlichen Konformationen einnehmen können, abseits des klassischen aktiven und inaktiven Zustands, die wiederum diverse Signalkaskaden auslösen können. Basierend auf dieser Trajektorie, verwenden die Studierenden die Programmiersprache Python, um die dynamischen Interaktionen zwischen dem Medikament und dem GPCR zu quantifizieren und in Form von Diagrammen zu visualisieren. Für viele war es ein Aha-Moment, als Interaktionen wie Wasserstoffbrücken und Salzbrücken, die sonst nur aus zweidimensionalen Strukturformeln bekannt waren, nun dreidimensional und über den zeitlichen Verlauf variierend gezeigt wurden.
In der dritten und letzten Woche fokussieren sich die Studierenden darauf, die Ergebnisse aus der Literaturrecherche und den Simulationen zusammenzutragen und in Form einer akademischen Arbeit zu konsolidieren.
Künftig spielt auch KI mit
Das Wahlpflichtpraktikum wird 2024/2025 durch den Innovationsfonds Lehre der FAU gefördert und soll
im kommenden Semester auch die Anwendung von künstlicher Intelligenz bei der Wirkstoffentwicklung
veranschaulichen. Interessierte Studierende sollen sich frühzeitig bei Eduard Neu (eduard.neu@fau.de) melden.
Literatur
[1] Klebe G. Wirkstoffdesign: Entwurf und Wirkung von Arzneistoffen. Berlin, Heidelberg, Springer Spektrum; 2023
[2] Manglik A et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016;537:185-190
[3] Fink EA et al. Structure-based discovery of nonopioid analgesics acting through the α2A-adrenergic receptor. Science 2022;377:eabn7065
[4] Rosenbaum DM et al. Structure and function of an irreversible agonist-β2 adrenoceptor complex. Nature 2011;469:236-240